ARTICLE
High-resolution, multicolor 3D imaging at the speed of life
An innovative laser fluorescence technique, swept confocally aligned planar excitation (SCAPE) microscopy,
overcomes the limitations of earlier methods and offers broad life sciences utility.
Overview
Researchers in diverse fields of life sciences have a common need for 3D fluorescence microscopy tools that are characterized by high speed, high pixel count, and single-cell resolution and can acquire images without causing significant photodamage to the specimen. Despite numerous developments and technical improvements, however, most established techniques still involve tradeoffs that compromise at least one of these parameters.

Figure 1: In SCAPE, a light sheet is formed at an oblique angle by off-axis illumination of the primary microscope objective with a line profile beam (a); SCAPE builds a volumetric image by scanning the light sheet while capturing a series of images of the illuminated plane (b).
Progress and tradeoffs
For example, confocal microscopy cannot image a large xyz volume in high resolution at multihertz repetition rates because of the speed limitations of physically scanning a single spot. Moreover, the short dwell time per pixel means that the fastest confocal scans require high laser power, resulting in significant photodamage to live samples.
And while two-photon microscopy dramatically reduces photodamage, such single-point approaches face the same problems resulting from speed/resolution/volume tradeoffs. Recently developed fast acousto-optic modulators (AOMs) now enable faster scanning of small pre-selected volumes, but this approach is of limited use for either large volumes or moving organisms.
Conventional light sheet microscopy enables an entire xy plane to be sampled simultaneously, but it requires lateral sample access (and therefore special preparation) as well as time to build up a cube of 3D data. In addition, synchronization of optics and stage motion make these techniques complex and slow.
Professor Elizabeth Hillman and her colleagues at Columbia University’s Zuckerman Mind Brain Behavior Institute (New York, NY) set out to develop an innovative approach that avoids these limitations while supporting a range of mounted and unmounted sample geometries. Their successful result is swept confocally aligned planar excitation (SCAPE) microscopy, which was first described in a 2015 publication.1 An updated version, SCAPE 2.0, was reported in 2019,2 and Leica Microsystems, recognizing its broad life sciences utility, has now licensed it.
How SCAPE works
Hillman explains, “We reasoned that really high-speed imaging can probably never come from single- or even multi-point scanning. Even if we could get the required scanning speed, the dwell time for each pixel would be too short to obtain images with acceptable signal-to-noise. So, we began thinking about light-sheet microscopy. Nearly all systems at that time needed two objectives positioned at 90° to each other around the sample. The question thus became, can we combine the multipixel advantages of a light sheet in a single-objective configuration?”
The team realized that using an off-axis path, through the edge of a high numerical-aperture objective, would enable creation of an excitation light sheet at, say, 45° to the true xy plane of the microscope (see Fig. 1). To image the fluorescence coming from this oblique plane, they rotated the imaging plane of the objective lens to focus the camera precisely, using an approach similar to oblique plane microscopy.3 Hillman and her team use a scanning mirror upstream of the objective to move the light sheet side to side, which also redirects returning fluorescence light to maintain focus on the moving light sheet. By stacking the planes as the mirror moves, the microscope can quickly and repeatedly generate images of 3D volumes.
Several details of SCAPE 2.0 (see Fig. 2) merit explanation. The problem of imaging a sloped plane (that is, a light sheet that is on an angle to the viewing axis) is addressed by relaying the captured fluorescence to form a real oblique image at an intermediate point using a second objective lens. This image is then captured through a second objective lens, arranged at an angle (approximately 127°) to focus the plane of the light sheet flat onto a camera.
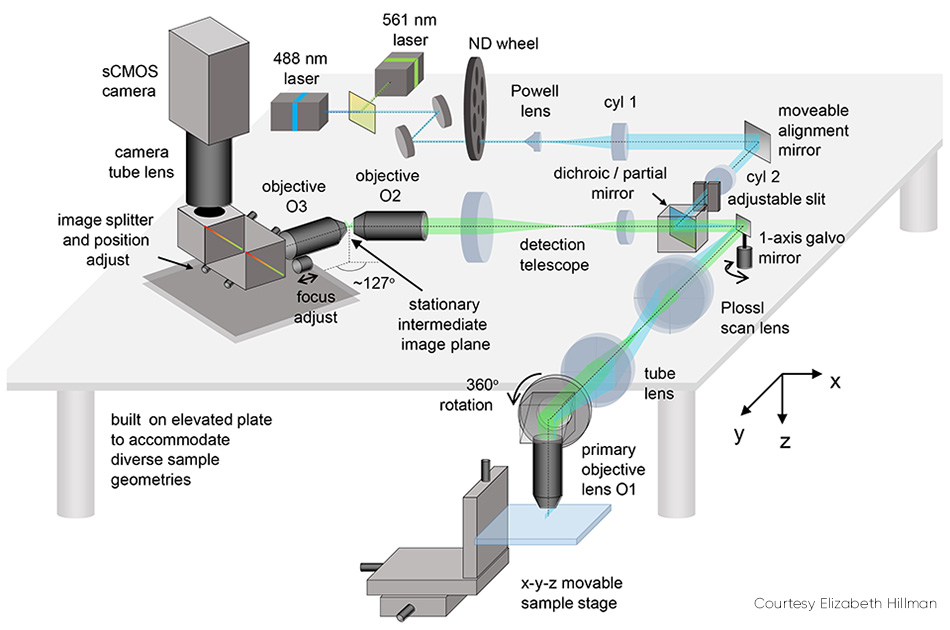
Figure 2: The movable alignment mirror is one of SCAPE 2.0’s key elements.
The final image on the camera is an oblique y-z plane from within the sample, which is usually a rectangle; it is narrower in the z direction (vs. y) because of the limited penetration of light into most tissues. In such samples, it is useful to operate the camera to only read out a reduced number of rows (corresponding to depths in z) since this permits even higher speed imaging—for example, 200 rows can be read out at between 1000 and 18,000 fps, depending on the camera used.
The issue of scan synchronization was first solved by scanning the light sheet using a polygon mirror. The detection path involved the facet adjacent to the one used by the excitation light. Hillman explains, “This polygon was the original inspiration for SCAPE, but we soon realized that it was simpler and just as effective to use a single galvanometer mirror. This change makes the system easier and cheaper to build, lets more light return to the camera, and makes it easier to control the system’s scan patterns.”
With no moving parts other than a galvo mirror, SCAPE’s overall speed is limited only by the camera frame rate and signal-to-noise ratio (SNR). Depending on the specific experiment, the galvo mirror is scanned at between 10 and 100 Hz, corresponding to an unprecedented 10–100 volumes per second (vps). SCAPE uses a conventional sawtooth scan pattern—that is, linear sweep followed by near-instantaneous reset. The amplitude of the galvo’s sweep, and the number of camera frames per sweep, determine the system’s field of view and sampling density in the x direction. Faster cameras can be leveraged to increase volume rates, sampling density, or field of view. While most of the team’s imaging has used standard sCMOS cameras, they reached over 300 vps imaging using an ultrafast CMOS camera with an integrated intensifier.
Because the light sheet is being swept at an angle to the image viewing axis z, each depth slice is slightly offset relative to the next slice. The microscope’s computer uses a simple transform to correct this ‘skew’ and generate an undistorted 3D image volume.
Digital laser modulation
Concurrent monitoring of multiple fluorophores, including functional indicators and fluorescent proteins, enables correlation of dynamic behavior (for example, muscle action) with molecular composition, cellular structure, neural signals, and so forth. SCAPE readily supports such applications by providing multiwavelength excitation—via plug-and-play Coherent OBIS optically pumped semiconductor lasers (OPSLs)—with the option to simultaneously acquire two or more spectrally separated images side by side on the camera.
Hillman cites several innovative advantages of OPSL technology for this work compared to earlier laser types. She notes the wide range of available wavelengths and power levels. “Years ago, we had 488 nm, 532 nm, and 638 nm, and that was about it if you wanted usable power levels. We had no options in the yellow and orange. But today, we can choose laser sources with tens and hundreds of milliwatts at wavelengths that closely match the excitation of almost any commonly available fluorophore.” She explains that most of their SCAPE systems integrate multiple free-space lasers, giving them more flexibility than fiber coupling. “It is super convenient that the lasers are compact and all have identical form factors and the same electronic interface.” To date, Hillman says, they have used as many as five laser wavelengths in some experiments. She also explains that she regularly takes SCAPE to workshops and courses and uses available OBIS lasers with minimal realignment.
Digital imaging is another important feature of OPSLs. Because the OPSLs can be turned on and off at speeds up to 25 kHz, excitation wavelength can be alternated on consecutive frames with precise timing. This is complemented by multiwavelength detection using a lab-built image-splitter composed of dichroic filters and mirrors. This device projects the spectrally separated images with fields of view up to 1280 voxels wide, with no effect on imaging speed compared to single-wavelength operation.
Demonstrating power and range
Two recent collaborative studies illustrate the power and range of SCAPE.
Imaging of small organisms—including the entirety of the body, brain, and nervous system—is a trend in neuroscience. Hillman and colleagues recently published a study describing their high-speed, 3D imaging of genetically encoded and calcium-sensitive fluorescent proteins in live Drosophila larvae (see Fig. 3). In addition to capturing the complex dynamics of the larva’s body and nervous system during peristaltic crawling, the team tracked how neurons along the body wall fired as they were deformed.
The team also used SCAPE to study dynamic firing of neuronal dendrites in the live rodent cortex5 and olfactory sensory neurons in the mouse nose,6 and to image entire, freely moving C. elegans worms. What’s more, they produced dramatic videos of beating, embryonic zebrafish hearts.2
Studies of the embryonic zebrafish heart can provide insights into vertebrate heart development, including the influence of genetic and environmental factors on structure and function. Conventional microscopy requires time gating that inevitably misses details such as irregular arrhythmias given the natural heart rate of 2 to 4 Hz, and it cannot perform full 4D particle tracking for red blood cell (RBC) flow analysis. Hillman’s team partnered with pediatric cardiologist Professor Kimara Targoff, whose lab uses zebrafish to decipher genetic mutations that can cause heart malformations in the embryo. The collaborative effort captured videos of both red blood cells coursing through the beating heart at over 100 vps, and leveraged GCaMP labeling to capture individual waves of calcium activity coursing across the beating heart (see Fig. 4).
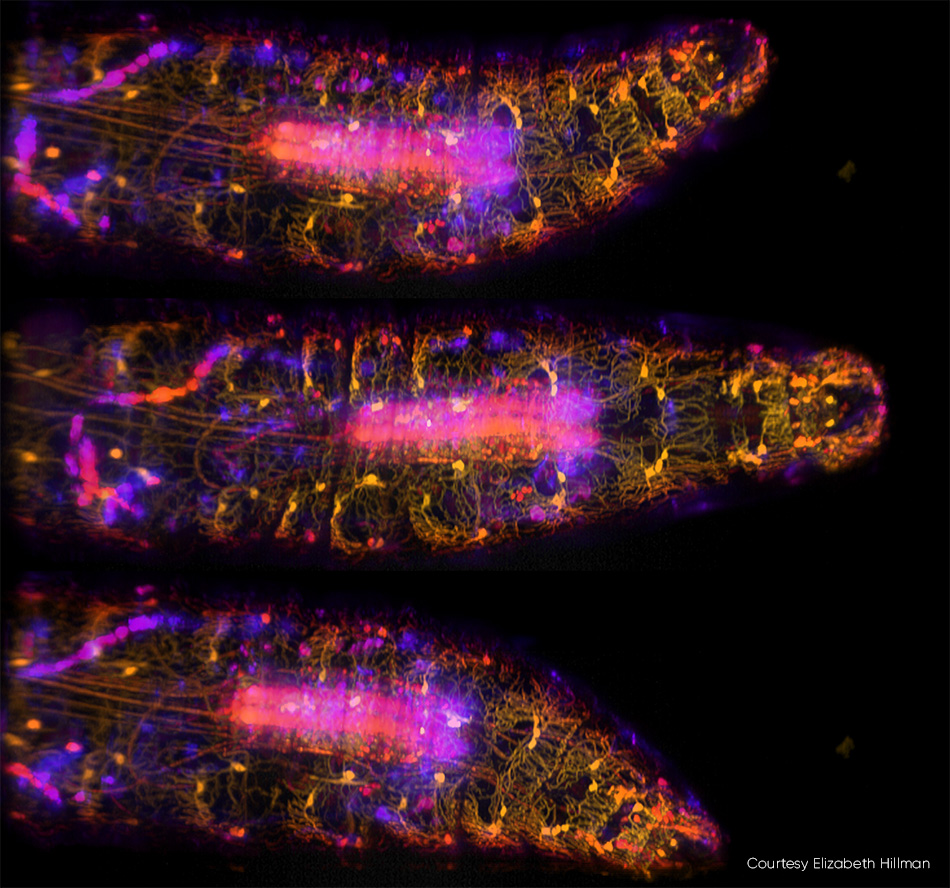
Figure 3: In these three images of a moving Drosophila larva captured by SCAPE 2.0 at 10 vps [3] ventral proprioceptive neurons are labeled with GFP and imaged using 488 nm excitation. Colors (from yellow to blue) denote signals from different depths into the sample. For full details, see R. Vaadia et al. [4] and for a real-time video sequence from this study, see http://bit.ly/SCAPE2019.
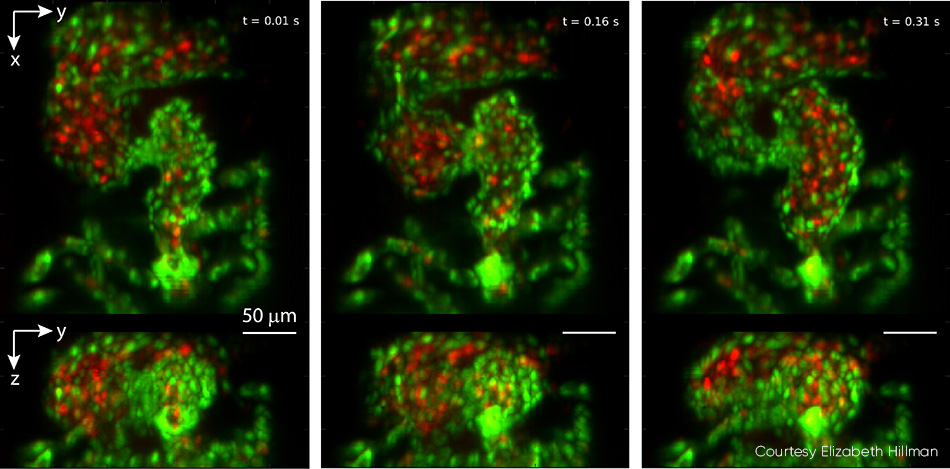
Figure 4: This triptych, drawn from video showing a zebrafish heart beating in real time, was captured at 100 vps. Top panels show z-projection and bottom panels show x-projection. The heart’s ventricle begins compressed, with the outflow valve open, and then fills from the atrium in successive images. Endothelial cells of the heart wall are labeled with EGFP (green) while red blood cells are labelled with DsRed (red). Both fluorophores were excited with 488 nm laser light (0.6 mW at the sample). For full details, including video, see V. Voleti et al. [2]
Summary
Across life sciences, fluorescence microscopy is used as a tool that allows researchers to connect events at the molecular, cellular, organ, and organism levels. The ability to record high-resolution multicolor (3D) images at the speed of life—4D microscopy—is now poised to play a key role in accelerating this research.
References
REFERENCES
1. M. B. Bouchard et al., Nat. Photonics, 9, 2, 113–119 (2015).
2. V. Voleti et al., Nat. Methods, 16, 10, 1054–1062 (2019).
3. C. Dunsby, Opt. Express, 16, 25, 20306–20316 (2008).
4. R. Vaadia et al., bioRxiv, 467274 (2018).
5. E. M. Hillman et al., Curr. Opin. Neurobiol., 50, 190–200 (2018).
6. L. Xu et al., Science, 368, 6487, eaaz5390 (2020).